scRNA-seq
Fundamentals of scRNA-seq analysis in Python Lecture 3, 01-28-2025¶
This notebook is part the course 580.447/647 Computational Stem Cell Biology taught by Patrick Cahan at Johns Hopkins University in Winter/Spring 2025. Many parts of this notebook are derived from the sources listed in the Resources section at the end of this notebook.
Objectives for today¶
After completing this tutorial, students will be familiar with:
- Basics of Scanpy
- annData
- loading and saving files
- getting var info
- getting more help: scanpy docs, tutorials, ChatGPT
- Quality control
- Normalization
- Dimension reduction
- Cell-cell distance
- Unsupervised clustering
- Differential gene expression
- Cell annotation
Background on the data¶
Let's start with some background + motivation for the data set that we are going to analyze: hematopoietic cells from peripheral blood.
Hematopoiesis¶
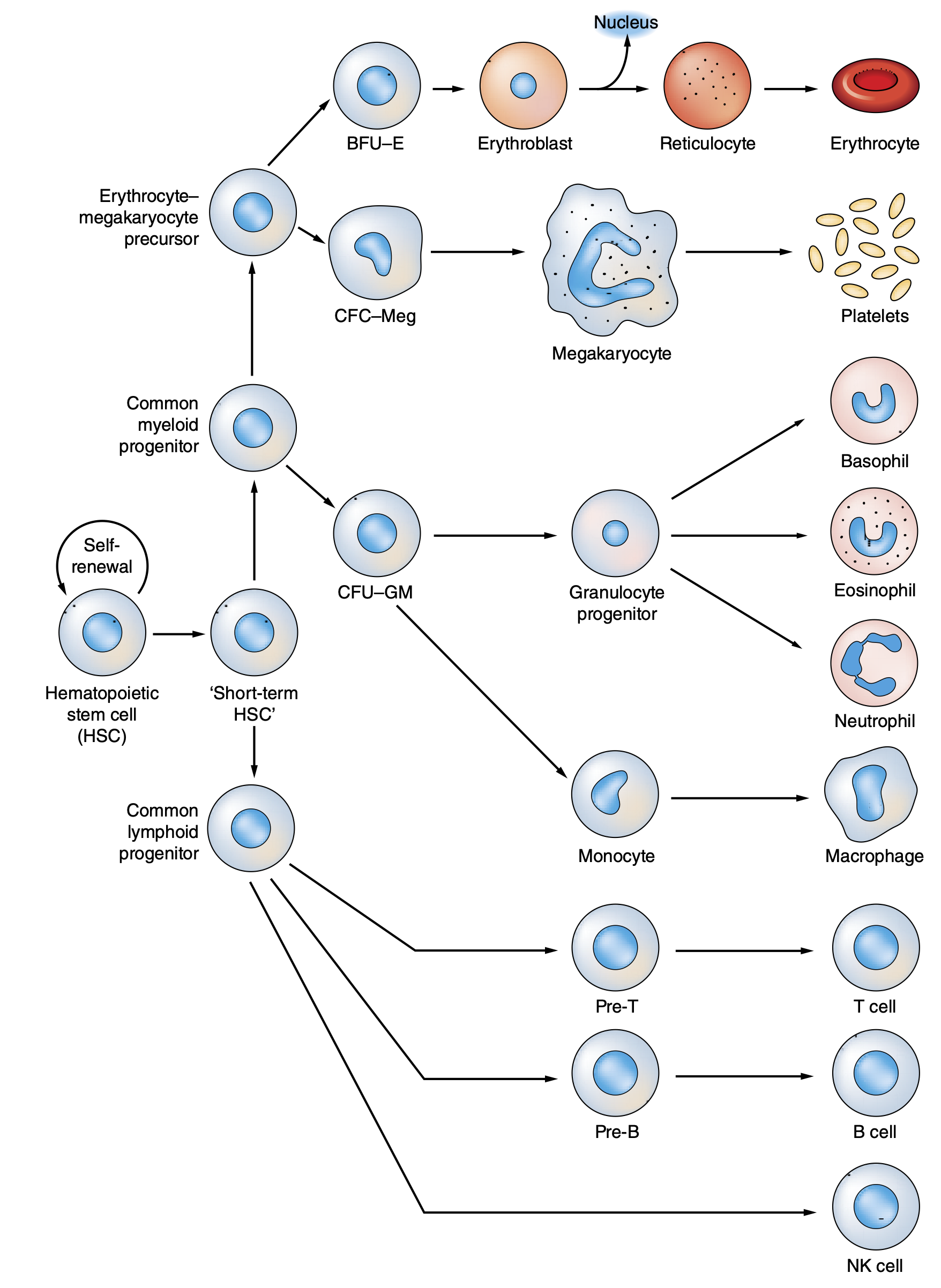
These cells are produced in the bone marrow and then move into the vascular so that they can move about the body and perform their functions (transport oxygen, clot, do adaptive and innate immune things).
Peripheral blood mononuclear cells (PBMCs)¶
It is possible to enrich for what are called PBMCs from vein by subjecting sample to density gradient centrifugation. This enrichs for cells that have one, round nucleus, and excludes cells that do not have nuclie, or that have multi-lobed structures. PBMCs typically contain:
... several classes of immune cells, including T cells (~70%), B cells (~15%), monocytes (~5%), dendritic cells (~1%) and natural killer (NK) cells (~10%)
-- Sen et al 2019
and
... The CD3+ lymphocytes are composed of CD4+ and CD8+ T cells, roughly in a 2:1 ratio.
-- Kleiveland, 2015
Bio question(s) for the day¶
- What does scRNAseq estimate as their relative proportion? and how does it compare to the estimate above?
- To what extent does cell type composition vary between samples?
- Are there other genes that are better at distinguishing between these populations than marker genes listed below?
- How many cells do you need to reliably detect a sub-population?
Data¶
10X Genomics scRNA-seq on human PBMCs
Sample 1¶
Sample 2¶
- https://colab.research.google.com/corgiredirector?site=https%3A%2F%2Fcf.10xgenomics.com%2Fsamples%2Fcell-exp%2F6.1.0%2F20k_PBMC_3p_HT_nextgem_Chromium_X%2F20k_PBMC_3p_HT_nextgem_Chromium_X_filtered_feature_bc_matrix.h5
- 20k Human PBMCs, 3’ HT v3.1, Chromium X
- Sourced from a healthy female donor
- 23,837 cells
- 35,000 reads per cell
- Filtered data in .h5 format
CellRanger¶
CellRanger is the program that converts sequencing reads into gene expression counts.
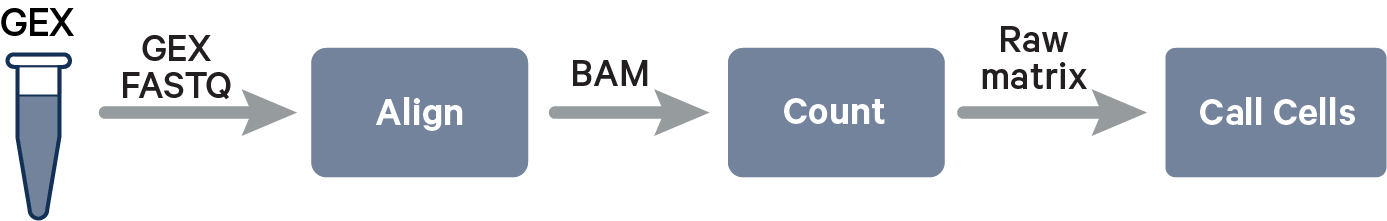
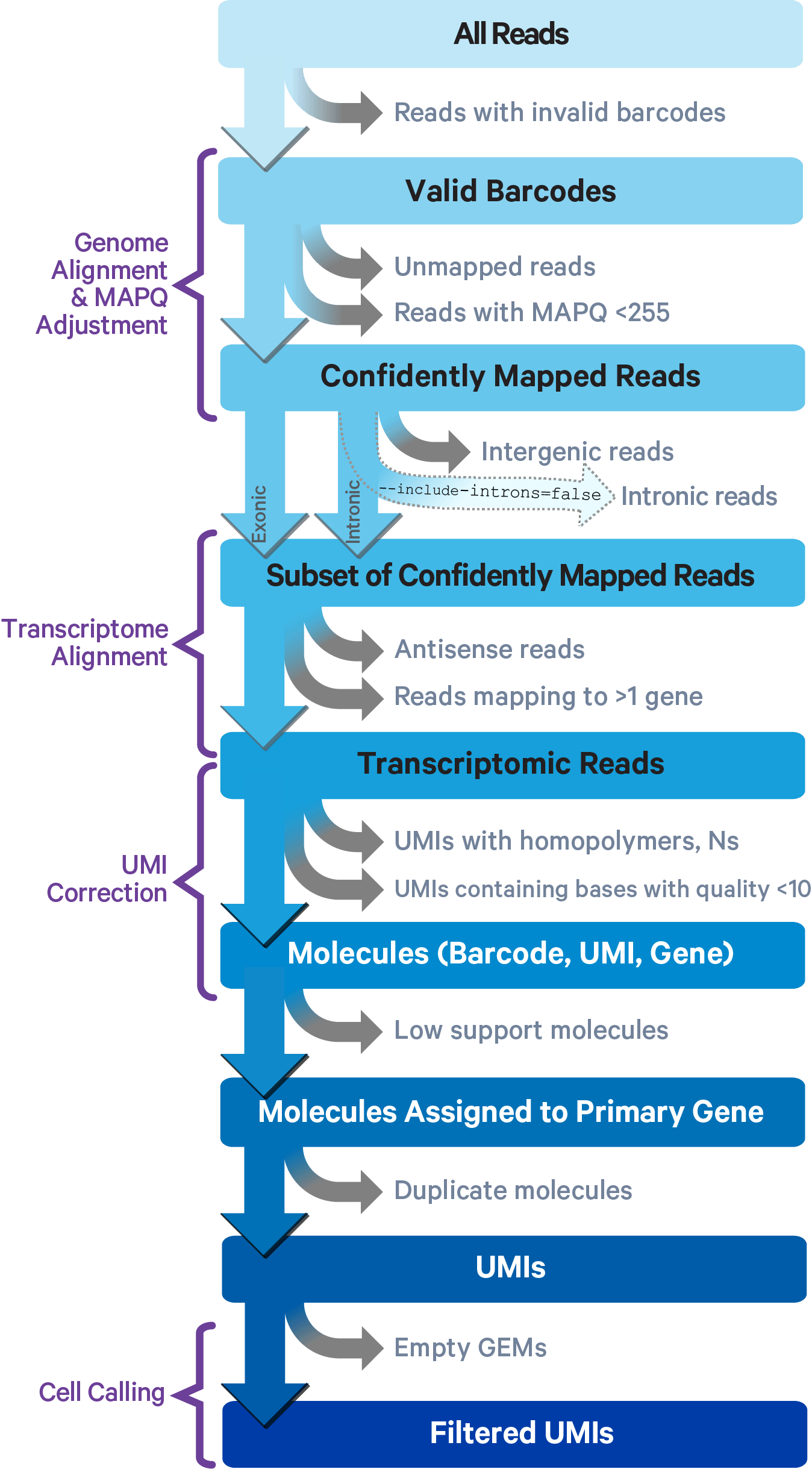
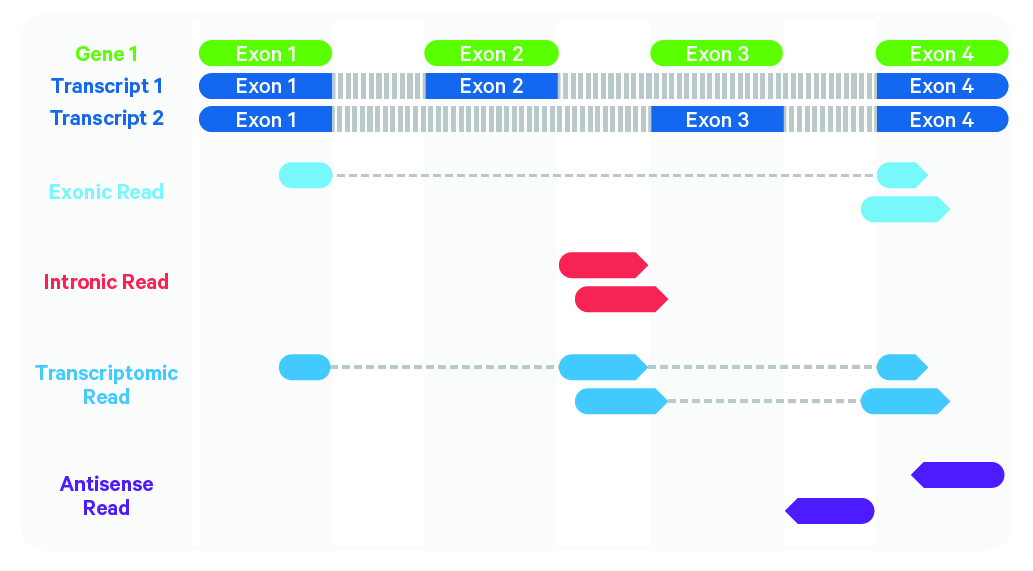
Here is a more detailed glimpse of what is happening here:
# !pip install scanpy scipy umap-learn leidenalg
Fetch data file(s)¶
# wget for linux/unix
# !wget https://cf.10xgenomics.com/samples/cell-exp/3.0.0/pbmc_10k_v3/pbmc_10k_v3_filtered_feature_bc_matrix.h5
# !wget https://cf.10xgenomics.com/samples/cell-exp/6.1.0/20k_PBMC_3p_HT_nextgem_Chromium_X/20k_PBMC_3p_HT_nextgem_Chromium_X_filtered_feature_bc_matrix.h5
# curl for macOS; i have no clue what to use for Windows
# !curl https://cf.10xgenomics.com/samples/cell-exp/3.0.0/pbmc_10k_v3/pbmc_10k_v3_filtered_feature_bc_matrix.h5 -o pbmc_10k_v3_filtered_feature_bc_matrix.h5
# !curl https://cf.10xgenomics.com/samples/cell-exp/6.1.0/20k_PBMC_3p_HT_nextgem_Chromium_X/20k_PBMC_3p_HT_nextgem_Chromium_X_filtered_feature_bc_matrix.h5 -o 20k_PBMC_3p_HT_nextgem_Chromium_X_filtered_feature_bc_matrix.h5
import scanpy as sc
import numpy as np
import pandas as pd
import matplotlib.pyplot as plt
import warnings
# warnings.filterwarnings('ignore')
plt.rcParams['figure.dpi'] = 300
sc.logging.print_header()
/opt/homebrew/Caskroom/miniforge/base/envs/scnpy/lib/python3.12/site-packages/tqdm/auto.py:21: TqdmWarning: IProgress not found. Please update jupyter and ipywidgets. See https://ipywidgets.readthedocs.io/en/stable/user_install.html from .autonotebook import tqdm as notebook_tqdm
scanpy==1.10.4 anndata==0.11.3 umap==0.5.7 numpy==1.26.4 scipy==1.14.1 pandas==2.2.3 scikit-learn==1.6.1 statsmodels==0.14.4 igraph==0.11.8 pynndescent==0.5.13
Load Sample 1¶
# I have stored these data files in another dir
ad10f = sc.read_10x_h5("data/pbmc_10k_v3_filtered_feature_bc_matrix.h5")
# If you stored yours in the cwd on Colab, you should uncomment and run this line instead to load your data:
# ad10f = sc.read_10x_h5("pbmc_10k_v3_filtered_feature_bc_matrix.h5")
/opt/homebrew/Caskroom/miniforge/base/envs/scnpy/lib/python3.12/site-packages/anndata/_core/anndata.py:1758: UserWarning: Variable names are not unique. To make them unique, call `.var_names_make_unique`.
utils.warn_names_duplicates("var")
/opt/homebrew/Caskroom/miniforge/base/envs/scnpy/lib/python3.12/site-packages/anndata/_core/anndata.py:1758: UserWarning: Variable names are not unique. To make them unique, call `.var_names_make_unique`.
utils.warn_names_duplicates("var")
Getting information about functions¶
You can get info about functions using
help(functionName)
or
?functionName
Inspecting the data -- gross properties¶
ad10f.var_names_make_unique()
#?sc.read_10x_h5
Package documentation¶
Often, the best source of help is the package's documentation.
print(ad10f)
AnnData object with n_obs × n_vars = 11769 × 33538
var: 'gene_ids', 'feature_types', 'genome'
ad10f.shape
(11769, 33538)
type(ad10f)
anndata._core.anndata.AnnData
AnnData¶
AnnData is a Python package that defines a data structure designed to efficiently store large data sets like scRNA-seq.

Slicing or subsetting¶
There are lots of ways to get parts of an adata obkect. Here, let's randomly select a subset of cells and genes.
ncells = 3
ngenes = 4
randGenes = np.random.choice(ad10f.var_names, size=ngenes, replace=False)
randCells = np.random.choice(ad10f.obs_names, size=ncells, replace=False)
print(randCells)
['TTCCACGCAGACGATG-1' 'CATTGCCAGGTACAAT-1' 'ACTTTCAGTTGAGAGC-1']
print(randGenes)
['AC130352.1' 'AC092354.2' 'MOAP1' 'POLR3B']
newAnndata = ad10f[ randCells ]
newAnndata
View of AnnData object with n_obs × n_vars = 3 × 33538
var: 'gene_ids', 'feature_types', 'genome'
ad10f[ randGenes ]
--------------------------------------------------------------------------- KeyError Traceback (most recent call last) Cell In[13], line 1 ----> 1 ad10f[ randGenes ] File /opt/homebrew/Caskroom/miniforge/base/envs/scnpy/lib/python3.12/site-packages/anndata/_core/anndata.py:1022, in AnnData.__getitem__(self, index) 1020 def __getitem__(self, index: Index) -> AnnData: 1021 """Returns a sliced view of the object.""" -> 1022 oidx, vidx = self._normalize_indices(index) 1023 return AnnData(self, oidx=oidx, vidx=vidx, asview=True) File /opt/homebrew/Caskroom/miniforge/base/envs/scnpy/lib/python3.12/site-packages/anndata/_core/anndata.py:1003, in AnnData._normalize_indices(self, index) 1002 def _normalize_indices(self, index: Index | None) -> tuple[slice, slice]: -> 1003 return _normalize_indices(index, self.obs_names, self.var_names) File /opt/homebrew/Caskroom/miniforge/base/envs/scnpy/lib/python3.12/site-packages/anndata/_core/index.py:32, in _normalize_indices(index, names0, names1) 30 index = tuple(i.values if isinstance(i, pd.Series) else i for i in index) 31 ax0, ax1 = unpack_index(index) ---> 32 ax0 = _normalize_index(ax0, names0) 33 ax1 = _normalize_index(ax1, names1) 34 return ax0, ax1 File /opt/homebrew/Caskroom/miniforge/base/envs/scnpy/lib/python3.12/site-packages/anndata/_core/index.py:105, in _normalize_index(indexer, index) 103 if np.any(positions < 0): 104 not_found = indexer[positions < 0] --> 105 raise KeyError( 106 f"Values {list(not_found)}, from {list(indexer)}, " 107 "are not valid obs/ var names or indices." 108 ) 109 return positions # np.ndarray[int] 110 raise IndexError(f"Unknown indexer {indexer!r} of type {type(indexer)}") KeyError: "Values ['AC130352.1', 'AC092354.2', 'MOAP1', 'POLR3B'], from ['AC130352.1', 'AC092354.2', 'MOAP1', 'POLR3B'], are not valid obs/ var names or indices."
ad10f[:, randGenes ]
View of AnnData object with n_obs × n_vars = 11769 × 4
var: 'gene_ids', 'feature_types', 'genome'
gname_counts = ad10f.var_names.value_counts()
print(gname_counts)
np.any(gname_counts>1)
MIR1302-2HG 1
DCT 1
GPC6-AS2 1
GPC6 1
AL354811.1 1
..
ORC3 1
RARS2 1
SLC35A1 1
AL049697.1 1
FAM231C 1
Name: count, Length: 33538, dtype: int64
False
duplicates = gname_counts[gname_counts > 1]
print(duplicates)
Series([], Name: count, dtype: int64)
ad10f.var_names_make_unique()
ad10f[:, randGenes ]
View of AnnData object with n_obs × n_vars = 11769 × 4
var: 'gene_ids', 'feature_types', 'genome'
ad10f[randCells, randGenes ]
View of AnnData object with n_obs × n_vars = 3 × 4
var: 'gene_ids', 'feature_types', 'genome'
Views are pointers to memory where original anndata is stored. They are not copies of the variable.
Gene counts¶
So, where is the data?
ad10f[randCells, randGenes ].X
<Compressed Sparse Row sparse matrix of dtype 'float32' with 1 stored elements and shape (3, 4)>
The Metadata¶
- for cells, see .obs
- for genesm see .var
print(ad10f.obs.columns)
Index([], dtype='object')
ad10f.obs['sample_name'] = "sample_1"
print(ad10f.obs.columns)
Index(['sample_name'], dtype='object')
print(ad10f[randCells].obs)
sample_name TTCCACGCAGACGATG-1 sample_1 CATTGCCAGGTACAAT-1 sample_1 ACTTTCAGTTGAGAGC-1 sample_1
print(ad10f.var.columns)
Index(['gene_ids', 'feature_types', 'genome'], dtype='object')
print(ad10f[:,randGenes].var)
gene_ids feature_types genome AC130352.1 ENSG00000270154 Gene Expression GRCh38 AC092354.2 ENSG00000272370 Gene Expression GRCh38 MOAP1 ENSG00000165943 Gene Expression GRCh38 POLR3B ENSG00000013503 Gene Expression GRCh38
.loc¶
.obs and .var are Pandas dataframes and so you can access subsets of them using .loc and .iloc
print(ad10f.var.loc[randGenes])
gene_ids feature_types genome AC130352.1 ENSG00000270154 Gene Expression GRCh38 AC092354.2 ENSG00000272370 Gene Expression GRCh38 MOAP1 ENSG00000165943 Gene Expression GRCh38 POLR3B ENSG00000013503 Gene Expression GRCh38
Quality Control (QC)¶
Broadly speaking there are two things that we apply quality control to:
- Cells: We want to find putative doublets and low quality cell barcodes and hold them out of downstram analysis.
- Genes: We also can remove genes that are not detected in vast majority of cell
The function sc.pp.calculate_qc_metrics() will calculate a set of metrics on adata and place the results in .obs or .var. The results can then be used to filter the adata. pp.calculate_qc_metrics() can also take inputs to specify other, custom metrics to compute.
Below, we use it to compute the fraction of all UMIs in each cell that are mitochondrially encoded or that encode for ribosomal genes.
ad10f.var['mt'] = ad10f.var_names.str.startswith('MT-')
ribo_prefix = ("RPS","RPL")
ad10f.var['ribo'] = ad10f.var_names.str.startswith(ribo_prefix)
sc.pp.calculate_qc_metrics(ad10f, qc_vars=['mt','ribo'], percent_top=None, log1p=False, inplace=True)
Visualize¶
Take a look some typical QC values across these data
adClean = ad10f.copy()
fig, (ax1, ax2, ax3) = plt.subplots(1, 3, figsize=(10,4), gridspec_kw={'wspace':0.25}, constrained_layout=True)
ax1_dict = sc.pl.scatter(adClean, x='total_counts', y='pct_counts_mt', ax=ax1, show=False)
ax2_dict = sc.pl.scatter(adClean, x='total_counts', y='n_genes_by_counts',ax=ax2, show=False)
ax3_dict = sc.pl.scatter(adClean, x='pct_counts_ribo', y='n_genes_by_counts',ax=ax3, show=False)
plt.show()

Now, do the filtering.
First, keep cells with fewer than 20% mitochondrially encoded gene total UMIs
adClean = adClean[adClean.obs['pct_counts_mt']<20,:].copy()
adClean.n_obs
11047
Second, filter based on total number of genes detected (at least 500). Third, filter based on total number of counts (fewer than than 30,000) Fourth, keep genes that are detected in at least 3 cells.
sc.pp.filter_cells(adClean, min_genes=500)
print(adClean.n_obs)
sc.pp.filter_cells(adClean, max_counts=30000)
print(adClean.n_obs)
sc.pp.filter_genes(adClean, min_cells=3)
print(adClean.shape)
10924 10891 (10891, 20181)
Normalization¶
Simple library size scaling, then log transform. There are many other ways to correct for cell-to-cell technical variation, for example different cell lysis efficiencies, but this method is good enough for this data set and our goals.
adNorm = adClean.copy()
adNorm.layers['counts'] = adNorm.X.copy()
sc.pp.normalize_total(adNorm , target_sum=1e4)
sc.pp.log1p(adNorm)
adNorm.layers['lognorm'] = adNorm.X.copy()
Highly variable genes (HVG)¶
It is common practice to limit some parts of analysis to those variables/genes that exhibit some degree of variation in their values across the data. We call these highly variable genes, or HVG for short. To find these, calculate some gene statistics, which when combined with thresholds below, determine which genes are considered HVG. The relevant metrics are:
- mean expression
- dispersion, which is the variance / mean.
Variance is defined as the expected squared deviation of gene expression. The normalized dispersion is calculated by scaling based on a bin of mean expression.
sc.pp.highly_variable_genes(adNorm , min_mean=0.0125, max_mean=6, min_disp=0.25)
adNorm.var
| gene_ids | feature_types | genome | mt | ribo | n_cells_by_counts | mean_counts | pct_dropout_by_counts | total_counts | n_cells | highly_variable | means | dispersions | dispersions_norm | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| AL627309.1 | ENSG00000238009 | Gene Expression | GRCh38 | False | False | 60 | 0.005183 | 99.490186 | 61.0 | 60 | False | 0.007519 | 0.595591 | -0.001919 |
| AL627309.3 | ENSG00000239945 | Gene Expression | GRCh38 | False | False | 4 | 0.000340 | 99.966012 | 4.0 | 4 | False | 0.000389 | 0.144284 | -1.366927 |
| AL669831.5 | ENSG00000237491 | Gene Expression | GRCh38 | False | False | 679 | 0.062367 | 94.230606 | 734.0 | 665 | False | 0.077585 | 0.586329 | -0.029932 |
| FAM87B | ENSG00000177757 | Gene Expression | GRCh38 | False | False | 13 | 0.001190 | 99.889540 | 14.0 | 13 | False | 0.001139 | 0.169543 | -1.290532 |
| LINC00115 | ENSG00000225880 | Gene Expression | GRCh38 | False | False | 350 | 0.031269 | 97.026085 | 368.0 | 337 | False | 0.038579 | 0.459107 | -0.414723 |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| AC011043.1 | ENSG00000276256 | Gene Expression | GRCh38 | False | False | 77 | 0.006882 | 99.345739 | 81.0 | 74 | False | 0.010262 | 0.738345 | 0.429851 |
| AL592183.1 | ENSG00000273748 | Gene Expression | GRCh38 | False | False | 32 | 0.002719 | 99.728099 | 32.0 | 30 | False | 0.003977 | 0.799225 | 0.613985 |
| AC007325.4 | ENSG00000278817 | Gene Expression | GRCh38 | False | False | 239 | 0.020902 | 97.969241 | 246.0 | 233 | False | 0.023936 | 0.329110 | -0.807909 |
| AL354822.1 | ENSG00000278384 | Gene Expression | GRCh38 | False | False | 319 | 0.028210 | 97.289489 | 332.0 | 303 | False | 0.038089 | 0.594378 | -0.005588 |
| AC240274.1 | ENSG00000271254 | Gene Expression | GRCh38 | False | False | 101 | 0.008752 | 99.141813 | 103.0 | 99 | False | 0.010213 | 0.301150 | -0.892475 |
20181 rows × 14 columns
sc.pl.highly_variable_genes(adNorm)

Dimensionality reduction with Principal components analysis (PCA)¶
Why performan dimentaionality reduction?
- Genes are expressed in coordinated fashion, meaning that many have correlated expression patterns and so PCA reduces the computational complexity of downstream analysis
- scRNA-seq data is noisy on a per gene basis.
PCA allows us to reduce a high dimensional data set into a lower dimension in which much of the total variation is maintained. To understand PCA, you need to know linear algebra. In essence, it identifies sets of linear combinations of genes in such a way that a PC is uncorrelated with other PCs and that explains most variation in the data. The function call sc.tl.pca does the following:
- computes the covariance matrix (correlation between each pair of genes)
- finds eigenvectors (directions of axes that maximize that variance), orthogonal to each other
Later, we select a threshold (n_pcs) as the number of PCs that contribute most to the total variation on our data. We use these n_pcs PCs to compute cell-to-cell distances for embedding (2D visualization via UMAP) and for clustering of cells.
sc.tl.pca(adNorm , mask_var='highly_variable')
Marker genes¶
Here PBMC cell types and some genes that have been used to identify them:
Monocytes: CD14, CD68, LYZ
Dendritic: CLEC4C, FLT3
NK cells: GNLY, NCAM1
B cells: CD19, CD79A, CD79B
T cells: CD3D, CD3E, TRAC, TRBC
markerGenes = ['CD14','CLEC4C', 'CD19', 'GNLY', 'CD3D']
sc.pl.pca(adNorm , color=markerGenes, ncols=2, layer='counts')

sc.pl.pca(adNorm , color=markerGenes, ncols=2)

sc.pl.pca(adNorm, color=['total_counts', 'pct_counts_mt', 'pct_counts_ribo'], s=25,ncols=3)

sc.pl.pca_variance_ratio(adNorm, 50)

PCA Loadings¶
Where are PCs (i.e. loadings -- the contribution of each gene to each PC) stored in anndata? .varm (see figure of anndata way above)?
np.shape(adNorm.varm["PCs"])
(20181, 50)
Where are the PC values of each cell (i.e. the PC scores) stored in anndata? .obsm
np.shape(adNorm.obsm["X_pca"])
(10891, 50)
kNN cell-cell distances¶
The k-nearest neighbor graph generates an adjacency matrix by finding, for each cell, the k cells that are closest to it. This helps to reduce noise in computing cell-cell distances or similarities and is important for embedding the cells in a 2D space, and for community detection algorithms. Two arguments are n_neighbors, or k, and the number of PCs to use when computing cell-cell distances.
n_neighbors = 20
n_pcs = 10
sc.pp.neighbors(adNorm, n_neighbors=n_neighbors, n_pcs=n_pcs)
Visualization of cell-cell similarities¶
UMAP embedding¶
This an elaboration on the t-SNE embedding approach. Both of these methods try to project cells into a reduced (typically 2) dimensional coordinate system while maintaining both the global and local structure of the high dimensional space of the data (i.e. cells that are distant from each other high dimensions should still be distant in the reduced embedding; same is true for cells that are near each other). The distances come from the kNN graph.
sc.tl.umap(adNorm, 0.25)
/var/folders/1_/v5grqr5n7kj42276bb6ryxj00000gn/T/ipykernel_66578/2021512077.py:1: FutureWarning: The specified parameters ('min_dist',) are no longer positional. Please specify them like `min_dist=0.25`
sc.tl.umap(adNorm, 0.25)
sc.pl.umap(adNorm, color=markerGenes, alpha=.75, s=15, ncols=2)

Unsupervised clustering¶
Let's try to assign cells to distinct groups or clusters based on the cell-to-cell distances. Leiden and Louvain are examples of community detection methods that perform this task by searching for the group assignment that maximize within group similarity (which is equivalent to within group edges of a knn graph) and to minimize between group similarity.
sc.tl.leiden(adNorm,.1)
/var/folders/1_/v5grqr5n7kj42276bb6ryxj00000gn/T/ipykernel_66578/3992796732.py:1: FutureWarning: In the future, the default backend for leiden will be igraph instead of leidenalg. To achieve the future defaults please pass: flavor="igraph" and n_iterations=2. directed must also be False to work with igraph's implementation. sc.tl.leiden(adNorm,.1)
sc.pl.umap(adNorm,color=['leiden'], alpha=.75, s=15, legend_loc='on data')

Embeddings do not always agree with cluster results! See cluster 2 above.
We can try to rectify this by using PAGA, which tries to infer dynamic relationships between clusters, to seed an initial embedding for UMAP that reflects overall cluster similarities.
In the next cell, we call the following functions
tl.paga() computes intercluster similarities
pl.paga() creates an intial embedding based on this
tl.umap() uses this embedding to start its optimization
Let's restart with the cleaned data.
adNorm = adClean.copy()
## Norm, HVG, PCA, kNN
sc.pp.normalize_total(adNorm , target_sum=1e4)
sc.pp.log1p(adNorm)
sc.pp.highly_variable_genes(adNorm , min_mean=0.0125, max_mean=6, min_disp=0.25)
sc.tl.pca(adNorm , use_highly_variable=True)
n_neighbors = 20
n_pcs = 10
sc.pp.neighbors(adNorm, n_neighbors=n_neighbors, n_pcs=n_pcs)
sc.tl.leiden(adNorm,.1)
sc.tl.paga(adNorm)
sc.pl.paga(adNorm, plot=False)
sc.tl.umap(adNorm, 0.25, init_pos='paga')
sc.pl.umap(adNorm,color=['leiden'], alpha=.75, s=15, legend_loc='on data')
/opt/homebrew/Caskroom/miniforge/base/envs/scnpy/lib/python3.12/site-packages/scanpy/preprocessing/_pca.py:374: FutureWarning: Argument `use_highly_variable` is deprecated, consider using the mask argument. Use_highly_variable=True can be called through mask_var="highly_variable". Use_highly_variable=False can be called through mask_var=None
warn(msg, FutureWarning)
/var/folders/1_/v5grqr5n7kj42276bb6ryxj00000gn/T/ipykernel_66578/3419511010.py:15: FutureWarning: The specified parameters ('min_dist',) are no longer positional. Please specify them like `min_dist=0.25`
sc.tl.umap(adNorm, 0.25, init_pos='paga')

sc.pl.umap(adNorm , color=markerGenes, ncols=2)

Make a figure that shows the embedding and expression of marker genes side-by-side. Note that I am adding markers for T cell, B cell, and Monocyte sub-types
marker_genes_broad_dict = {
'B cell': ['CD79A', 'PAX5'],
'Dendritic': ['CLEC4C', 'FCER1A'],
'Monocyte': ['CSF1R', 'FCGR3A'],
'NK cell': ['NKG7', 'GNLY'],
'T cell': ['TRAC', 'CD3E'],
}
fig, (ax1, ax2) = plt.subplots(1, 2, figsize=(10,4), gridspec_kw={'wspace':0.25})
ax1_dict = sc.pl.umap(adNorm,color=['leiden'], alpha=.75, s=10, legend_loc='on data', ax=ax1, show=False)
ax2_dict = sc.pl.dotplot(adNorm, marker_genes_broad_dict, 'leiden', dendrogram=True,ax=ax2, show=False)
plt.show()
WARNING: dendrogram data not found (using key=dendrogram_leiden). Running `sc.tl.dendrogram` with default parameters. For fine tuning it is recommended to run `sc.tl.dendrogram` independently. WARNING: Groups are not reordered because the `groupby` categories and the `var_group_labels` are different. categories: 0, 1, 2, etc. var_group_labels: B cell, Dendritic, Monocyte, etc.

Cluster annotation¶
We can walk down the rows of the dotplot and guess at the cell types of some of these clusters based on the specificty of marker expression:
| cluster | markers of | cell type |
|---|---|---|
| 5 | B, Dendritic, Mono | ??? |
| 1 | Monocyte | Monocyte |
| 4 | Monocyte, T | ??? |
| 3 | B | B-cell |
| 6 | Dendritic (B/Mono?) | Dendritic cell |
| 7 | none | Mystery cell |
| 0 | T cell | T cell |
| 2 | NK/T | ??? |
Some clusters are ambiguous because they express markers of more than one cell type. This could be due to the cluster containing > 1 cell type, or because the cell barcodes are doublets. If the former, then we should be able to sub cluster them. Let's try this.
Clusters to refine are 5, 4, and 2.
sc.tl.leiden(adNorm,.1, restrict_to=["leiden",["5"]])
sc.tl.dendrogram(adNorm, "leiden_R")
fig, (ax1, ax2) = plt.subplots(1, 2, figsize=(10,4), gridspec_kw={'wspace':0.25})
ax1_dict = sc.pl.umap(adNorm,color=['leiden_R'], alpha=.75, s=10, legend_loc='on data', ax=ax1, show=False)
ax2_dict = sc.pl.dotplot(adNorm, marker_genes_broad_dict, 'leiden_R', dendrogram=True,ax=ax2, show=False)
plt.show()
WARNING: Groups are not reordered because the `groupby` categories and the `var_group_labels` are different. categories: 0, 1, 2, etc. var_group_labels: B cell, Dendritic, Monocyte, etc.

Cluster 5 is split into 2 clusters now. Both of these sub-clusters express the Monocyte marker CD14, but are distinguished by their expression of dendritic marker FCER1A and the B cell genes CD79A and PAX5. Let's look at the QC metrics to see if that can give us additional hints as to whether these are real cells or are doublets.
sc.pl.violin(adNorm, ['total_counts'], groupby='leiden_R' )

Cluster 5 (and each sub-cluster) has a high number of Total counts. So let's annotate these as 'doublet'.
What about the other questionable clusters 4 and 2? Can we refine those?
# cluster 2 first
sc.tl.leiden(adNorm,.05, restrict_to=["leiden",["2"]])
sc.tl.dendrogram(adNorm, "leiden_R")
fig, (ax1, ax2) = plt.subplots(1, 2, figsize=(10,4), gridspec_kw={'wspace':0.25})
ax1_dict = sc.pl.umap(adNorm,color=['leiden_R'], alpha=.75, s=10, legend_loc='on data', ax=ax1, show=False)
ax2_dict = sc.pl.dotplot(adNorm, marker_genes_broad_dict, 'leiden_R', dendrogram=True,ax=ax2, show=False)
plt.show()
WARNING: Groups are not reordered because the `groupby` categories and the `var_group_labels` are different. categories: 0, 1, 2,0, etc. var_group_labels: B cell, Dendritic, Monocyte, etc.

It looks like 2,0 is T cell, and 2,1 is Natural Killer cell. Note that 2,0 still has a distinct profile as compared to 0.
What about cluster 4? And this time, when we do the sub-clustering, let's keep the way that we split up cluster 2.
sc.tl.leiden(adNorm,.1, restrict_to=["leiden_R",["4"]])
sc.tl.dendrogram(adNorm, "leiden_R")
fig, (ax1, ax2) = plt.subplots(1, 2, figsize=(10,4), gridspec_kw={'wspace':0.25})
ax1_dict = sc.pl.umap(adNorm,color=['leiden_R'], alpha=.75, s=10, legend_loc='on data', ax=ax1, show=False)
ax2_dict = sc.pl.dotplot(adNorm, marker_genes_broad_dict, 'leiden_R', dendrogram=True,ax=ax2, show=False)
plt.show()
WARNING: Groups are not reordered because the `groupby` categories and the `var_group_labels` are different. categories: 0, 1, 2,0, etc. var_group_labels: B cell, Dendritic, Monocyte, etc.

Is 4,1 multiplet?
sc.pl.violin(adNorm, ['total_counts', 'n_genes_by_counts'], groupby='leiden_R' )

Yes, it looks like 4,1 is similar in these QC profiles to cluster 5.
So our updated annotation table is now:
| cluster | markers of | cell type |
|---|---|---|
| 5 | B, Dendritic, Mono | doublet |
| 1 | Monocyte | Monocyte |
| 4,0 | Monocyte | Monocyte |
| 3 | B | B-cell |
| 6 | Dendritic (B/Mono?) | Dendritic cell |
| 7 | none | Mystery cell |
| 0 | T cell | T cell |
| 2,0 | T cell | T cell |
| 2,1 | NK cell | NK cell |
Why do we have two Monocyte clusters and two T cells clusters?
marker_genes_sub_dict = {
'B cell': ['CD79A'],
'Dendritic': ['CLEC4C'],
'CD14 monocyte': ['CD14'],
'CD16 monocyte': ['FCGR3A'],
'NK cell': ['GNLY'],
'CD4 T cell': ['CD4', 'GATA3'],
'CD8 T cell': ['CD8A', 'EOMES']
}
fig, (ax1, ax2) = plt.subplots(1, 2, figsize=(10,4), gridspec_kw={'wspace':0.25})
ax1_dict = sc.pl.umap(adNorm,color=['leiden_R'], alpha=.75, s=10, legend_loc='on data', ax=ax1, show=False)
ax2_dict = sc.pl.dotplot(adNorm, marker_genes_sub_dict, 'leiden_R', dendrogram=True,ax=ax2, show=False)
plt.show()
WARNING: Groups are not reordered because the `groupby` categories and the `var_group_labels` are different. categories: 0, 1, 2,0, etc. var_group_labels: B cell, Dendritic, CD14 monocyte, etc.

We are close to a final annotation now:
| cluster | markers of | cell type |
|---|---|---|
| 5 | B, Dendritic, Mono | doublet |
| 1 | Monocyte | CD14 Monocyte |
| 4,0 | Monocyte | CD16 Monocyte |
| 3 | B | B-cell |
| 6 | Dendritic (B/Mono?) | Dendritic cell |
| 7 | none | Mystery cell |
| 0 | T cell | CD4 T cell |
| 2,0 | T cell | CD8 T cell |
| 2,1 | NK cell | NK cell |
But what is cluster 7? It has low counts. If it is just ambient 'soup', then we would not expect it to have high levels of cell type specific genes. Let's do differential gene expression analysis to see what is upregulated in this cluster.
Differential gene expression¶
sc.tl.rank_genes_groups(adNorm, use_raw=False, groupby="leiden_R")
sc.pl.rank_genes_groups_dotplot(adNorm, n_genes=5, groupby="leiden_R", dendrogram=True, key='rank_genes_groups')

sc.pl.rank_genes_groups_dotplot(adNorm, n_genes=20, groupby="leiden_R", dendrogram=True, key='rank_genes_groups', groups=['7'])
WARNING: Groups are not reordered because the `groupby` categories and the `var_group_labels` are different. categories: 0, 1, 2,0, etc. var_group_labels: 7

Although this result could be refined and explored a lot, it is sufficient for us to address our question about the identity of cluster 7 as follows:
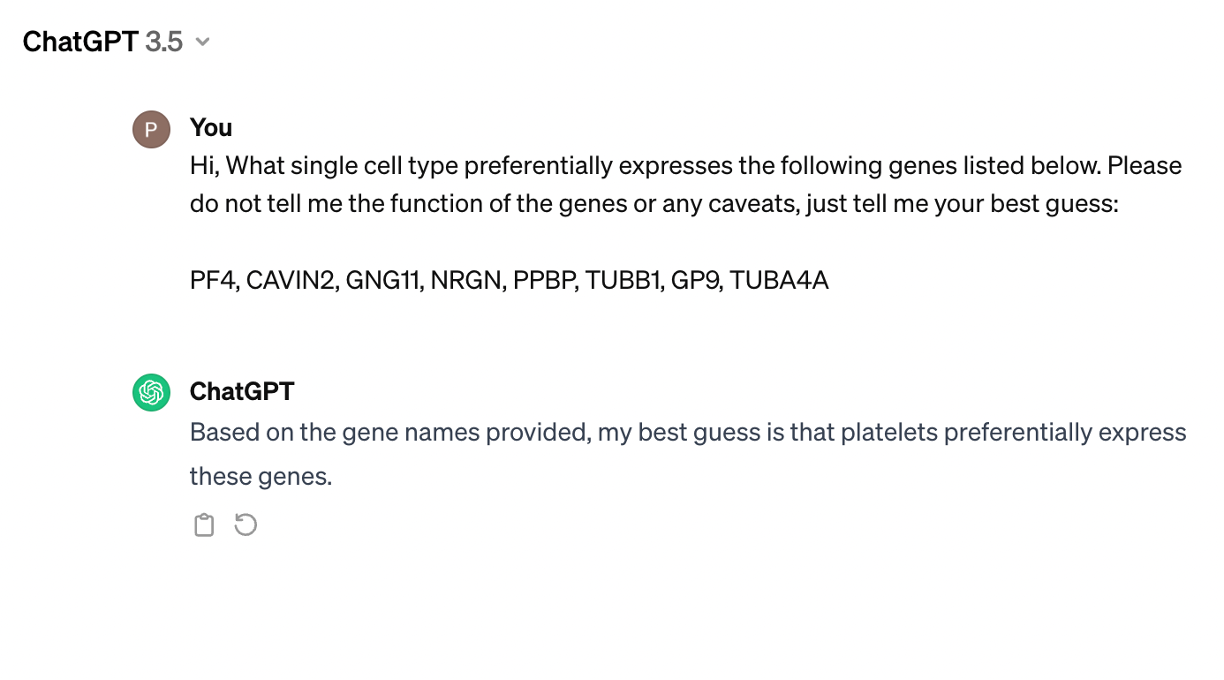
Now, let's add the annotation to the anndata.obs and discard the other cells
| cluster | markers of | cell type |
|---|---|---|
| 1 | Monocyte | CD14 Monocyte |
| 4,0 | Monocyte | CD16 Monocyte |
| 3 | B | B-cell |
| 6 | Dendritic (B/Mono?) | Dendritic cell |
| 7 | none | Platelet |
| 0 | T cell | CD4 T cell |
| 2,0 | T cell | CD8 T cell |
| 2,1 | NK cell | NK cell |
tokeep = ["1", "4,0", "3", "6", "7", "0", "2,0", "2,1"]
adNorm2 = adNorm.copy()
adNorm2 = adNorm2[adNorm2.obs['leiden_R'].isin(tokeep)].copy()
adNorm2.shape
adNorm2.obs['leiden_R'].value_counts()
leiden_R 0 3563 1 3115 3 1604 2,0 1031 2,1 611 4,0 368 6 81 7 60 Name: count, dtype: int64
cell_dict = {'Dendritic': ['6'],
'CD14 Monocyte': ['1'],
'CD16 Monocyte': ["4,0"],
'B cell': ['3'],
'Platelet': ['7'],
'CD4 T cell': ['0'],
'CD8 T cell': ["2,0"],
'NK cell': ["2,1"]
}
marker_genes_dict = {
'B cell': ['CD79A'],
'Dendritic': ['CLEC4C'],
'CD14 monocyte': ['CD14'],
'CD16 monocyte': ['FCGR3A'],
'NK cell': ['GNLY'],
'CD4 T cell': ['CD4', 'GATA3'],
'CD8 T cell': ['CD8A', 'EOMES'],
'Platelet': ['PF4', 'CAVIN2']
}
new_obs_name = 'cell_type'
adNorm2.obs[new_obs_name] = np.nan
for i in cell_dict.keys():
ind = pd.Series(adNorm2.obs.leiden_R).isin(cell_dict[i])
adNorm2.obs.loc[ind,new_obs_name] = i
adNorm2.obs['cell_type'] = adNorm2.obs['cell_type'].astype("category")
/var/folders/1_/v5grqr5n7kj42276bb6ryxj00000gn/T/ipykernel_66578/4042500018.py:28: FutureWarning: Setting an item of incompatible dtype is deprecated and will raise an error in a future version of pandas. Value 'Dendritic' has dtype incompatible with float64, please explicitly cast to a compatible dtype first. adNorm2.obs.loc[ind,new_obs_name] = i
sc.tl.dendrogram(adNorm2, "cell_type")
fig, (ax1, ax2) = plt.subplots(1, 2, figsize=(12,5), gridspec_kw={'wspace':0.4})
ax1_dict = sc.pl.umap(adNorm2,color=['cell_type'], alpha=.75, s=10, legend_loc='on data', ax=ax1, show=False)
ax2_dict = sc.pl.dotplot(adNorm2, marker_genes_dict, 'cell_type', dendrogram=True,ax=ax2, show=False)
plt.show()
WARNING: Groups are not reordered because the `groupby` categories and the `var_group_labels` are different. categories: B cell, CD14 Monocyte, CD16 Monocyte, etc. var_group_labels: B cell, Dendritic, CD14 monocyte, etc.

sc.pl.pca(adNorm2, color=['cell_type', 'GNLY'], alpha=.75, s=15, projection='3d')

Differential expression analysis¶
Let's identify genes that are preferenntially expressed in each cluster versus all the others as a final step to annotate these clusters and to remove ones that are likely to be doublets.
sc.tl.rank_genes_groups(adNorm2, use_raw=False, groupby="cell_type")
Dot plot of differentially expressed genes¶
First, we will apply a filter so that we only display genes that meet additional criteria beyond statistical: fold change, % expressed in cluster, % expressed in other cells
sc.tl.filter_rank_genes_groups(adNorm2, min_fold_change=.7, min_in_group_fraction=.5, max_out_group_fraction=.15)
sc.pl.rank_genes_groups_dotplot(adNorm2, n_genes=6, groupby="cell_type", dendrogram=True, key='rank_genes_groups_filtered')
